
Down Syndrome (Trisomy 21)
Sindrom Down atau Down Syndrome adalah kelainan kromosom yang utamanya menyebabkan defisit mental yang berasal dari genetik. Penyaringan dan pengelolaannya telah berkembang pesat. Tes apa yang saat ini dilakukan pada wanita hamil? Kapan amniosentesis (prosedur saat hamil untuk test air ketimbang) dilakukan? Penyebab, pemeriksaan, gejala, pengobatan…
Ini adalah kasus khusus aneuploidi, yang umumnya kromosom berpasangan (23 pasang pada manusia). Dalam kasus trisomi, setidaknya salah satu pasangannya adalah triplet, oleh karena itu dinamakan “trisomi”.
Ini dapat terjadi jika salah satu orang tua membawa 2 kromosom, bukan satu; fenomena ini juga dapat terjadi karena distribusi yang buruk selama pembelahan sel pertama, yang merupakan kasus paling sering, atau selama pembelahan sel kedua atau selanjutnya. Sel-sel individu dalam hal ini akan menjadi campuran sel sehat dan sel dengan sindrom Down. Kami berbicara dalam kasus trisomi mosaik ini. Sebaliknya, jika semua sel terpengaruh, kita berbicara tentang trisomi homogen.
Trisomi dikatakan bebas jika kromosom terlepas satu sama lain; jika terjadi translokasi, kromosom ekstra melekat pada yang lain.
Istilah “Down Syndrome” umumnya digunakan untuk merujuk pada orang dengan trisomi 21, atau sindrom Down, tetapi trisomi dapat melibatkan pasangan kromosom mana pun. Bergantung pada kromosom yang bersangkutan, dan jenis trisomi (mosaik atau tidak), individu yang terkena dapat bertahan hidup atau tidak.
Pada manusia, kebanyakan trisomi menyebabkan aborsi alami (keguguran), namun ada juga yang mengakibatkan kelahiran anak hidup-hidup.
Trisomi 21 – kadang-kadang disebut Down Syndrome – ditemukan pada tahun 1959 oleh sekelompok dokter Prancis: Marthe Gautier, Jérôme Lejeune, dan Raymond Turpin. Istilah “trisomi” menekankan pada kelainan genetik dengan 3 kromosom – tri – bukannya dua pada kromosom 21, maka dinamakan trisomi 21.
Gejala Down Syndrome
Trisomi 21 atau Down Syndrome ditandai dengan gejala-gejala berikut:
- keterbelakangan mental;
- wajah bulat;
- hidung kecil;
- gangguan kognitif yang kurang lebih signifikan;
- satu lipatan telapak tangan;
- mata sipit;
- leher yang rata;
- tangan lebar dan jari pendek;
- hipotonia (kurangnya tonus otot);
- satu ukuran lebih kecil;
- pertumbuhan lebih lambat;
- kemandulan.
Tips pencegahan Down Syndrome
Tidak ada cara untuk mencegah timbulnya sindrom Down.
Diagnosa Down Syndrome
Skrining gabungan untuk trisomi 21 pada trimester pertama kehamilan ditawarkan untuk semua wanita hamil. Ini terkait dengan trimester pertama kehamilan:
pengukuran ultrasonografi kejelasan leher janin;
tes penanda serum ibu.
Dokter kandungan, sonografer, dan ahli biologi akan menghubungi untuk pemeriksaan ini.
Jika hasil pemeriksaan ini tidak normal, risiko sindrom Down dinilai. Jika risiko ini tinggi (lebih dari atau sama dengan 1/250), dilakukan amniosentesis. Ini terdiri dari pengambilan cairan ketuban yang memungkinkan untuk menentukan kariotipe janin dan oleh karena itu untuk menegaskan atau menghilangkan, dengan pasti adanya trisomi 21. Praktik amniosentesis terbatas karena merupakan pemeriksaan. invasif, dengan risiko infeksi dan keguguran.
Mengembangkan metode skrining (test) Down Syndrome
Tes skrining genetik baru mencari, dari sampel darah yang diambil dari ibu, representasi DNA janin yang berlebihan dari kromosom 21 dalam DNA janin bebas yang beredar di dalam darah ibu. Sejak Januari 2019, tes ini telah ditanggung oleh Jamsostek:
- tes DNA bersirkulasi bebas untuk T21 ditawarkan, setelah skrining gabungan trimester pertama, kepada wanita yang perkiraan tingkat risikonya antara 1 dari 1000 dan 1 dari 51;
- untuk wanita yang risikonya lebih besar dari atau sama dengan 1 dari 50, rekomendasinya adalah melakukan amnocientesis.
- Amniosentesis masih diperlukan untuk memastikan diagnosis secara definitif, tetapi pemeriksaan invasif ini akan ditawarkan kepada lebih sedikit wanita (jumlah kariotipe janin akan dibagi empat), meskipun tingkat deteksi untuk trisomi 21 akan meningkat sekitar 15% berkat kinerja tes genetik.
Ketiga jenis sindrom Down
Penelitian telah menemukan bahwa kelainan kromosom akibat suatu nondisjungsi dapat muncul dalam tiga bentuk berbeda yang menimbulkan tiga jenis sindrom Down, yaitu trisomi bebas, trisomi mozaik, dan trisomi melalui translokasi.
1. Trisomi 21
Trisomi 21mempengaruhi sekitar 95% orang dengan trisomi 21. Ini adalah hasil dari kesalahan dalam distribusi kromosom, yang terjadi selama pembelahan sel pertama. Ini mempengaruhi semua sel dalam tubuh manusia.
2. Trisomi mosaik
Trisomi mosaik terjadi selama pembelahan sel kedua dan mempengaruhi sekitar 2% populasi penderita trisomi 21. Dalam hal ini, terdapat kombinasi sel yang memiliki 46 kromosom dan sel yang memiliki 47 kromosom.
Orang dengan trisomi mosaik terkadang memiliki perbedaan dibandingkan dengan orang lain dengan trisomi gratis: ciri fisik yang kurang jelas, lebih banyak potensi kognitif, dll. Namun, harus dihindari untuk menyimpulkan bahwa orang-orang ini tidak akan terlalu terpengaruh oleh trisomi, karena tidak mungkin untuk mengetahui sel mana yang memiliki kromosom ketiga.
3. Trisomi dengan translokasi
Pada tipe ini, ada sekitar 3% pengidap Down Syndrome.
Trisomi translokasi berarti bagian dari kromosom 21 telah rusak, dalam hal ini, anak menerima kromosom translokasi ini dalam susunan genetiknya dari salah satu orang tua, yang merupakan karier, meskipun ia tidak terpengaruh oleh sindrom tersebut.
Pertemuan dengan ahli genetika akan memverifikasi risiko melahirkan anak lain dengan sindrom Down.
Pengobatan Down Syndrome
Tidak ada pengobatan yang dapat meningkatkan kapasitas intelektual orang dengan sindrom Down. Di sisi lain, kualitas tindak lanjut medis dan kualitas pendidikan merupakan faktor penting dalam perkembangan dan kemandirian orang dengan sindrom Down.
Tindak lanjut medis yang baik sejak masa kanak-kanak adalah penting. “Pemantauan medis oleh profesional kesehatan yang berkualifikasi karenanya direkomendasikan sepanjang hidup” dapat dibaca di situs web Association Perce-Neige, yang menangani orang-orang cacat. Tindak lanjut awal diatur oleh tim multidisiplin dengan fisioterapis, terapis wicara. Anak-anak dengan sindrom Down sering bersekolah di sekolah luar biasa, meskipun ada juga contoh yang berhasil dari anak-anak dengan sindrom Down yang bersekolah di sekolah umum. Saat ini, banyak orang dengan sindrom Down bekerja, seringkali setelah pelatihan manual.
Harapan hidup rata-rata adalah lebih dari 50 tahun, tetapi kemajuan dalam kedokteran meningkatkan harapan hidup itu.
Kesehatan
Selain gangguan perkembangan kognitif, orang dengan sindrom Down sering kali memiliki beragam cacat lahir seperti penyakit jantung. Namun, harapan hidup – dan hidup sehat – telah meningkat pesat sejak masalah jantung dan paru-paru pertama kali dikendalikan sejak masa kanak-kanak.
Ada juga kelainan muskuloskeletal pada orang dengan sindrom Down yang dapat menyebabkan masalah tertentu. Hampir 30% populasi dengan sindrom Down memiliki salah satu kelainan berikut: dislokasi, displasia asetabular, hiperlaksitas sendi, hipotonia otot, ketidakstabilan sendi, kurangnya kontrol atas gerakan sukarela, dan keseimbangan statis dan dinamis.
Berkat kemajuan dalam pengobatan, gaya hidup yang lebih baik, dan perawatan pencegahan, kita sekarang menyaksikan semakin banyak anak-anak dengan sindrom Down berubah menjadi status remaja dan dewasa. Akibatnya, masalah yang terkait dengan penuaan yang memengaruhi pelanggan T21 telah menjadi, selama bertahun-tahun, masalah utama bagi organisasi.
Cacat dan perkembangan intelektual
Efek perkembangan sindrom Down bervariasi dari orang ke orang. Meskipun gangguan tersebut terkadang bisa parah, orang dengan karier memiliki kecacatan intelektual yang biasanya berkisar dari ringan hingga sedang (WHO menilai IQ antara 50 dan 69 sebagai keterbelakangan ringan). Oleh karena itu, dengan stimulasi dan sumber daya yang tepat, sebagian besar dari orang-orang ini mampu berintegrasi ke dalam masyarakat dengan sendirinya setelah dewasa.
Pengalaman telah menunjukkan bahwa mungkin untuk melakukan intervensi pada usia dini melalui stimulasi dini dan dengan demikian membantu perkembangan motorik, perkembangan intelektual dan perkembangan bahasa. Merangsang perkembangan anak dengan sindrom Down tidak jauh berbeda dengan merangsang anak lain. Ini tentang :
- untuk mendorongnya agar percaya diri;
- untuk mendorongnya membuat rencana;
- mendukung pengembangan keterampilan sosial;
- mendukung pengembangan keterampilan sosial;
- untuk mengenali kekuatannya dan membuatnya bangga atas prestasinya;
- untuk menamai kemajuannya;
- dengarkan sudut pandangnya.
Selain keterlibatan orang tua, nasihat ahli akan membantu mendukung perkembangan anak down syndrome, sehingga mereka bisa tumbuh dan berkembang seperti anak lainnya.
Adapun pendidikan anak-anak dengan sindrom Down, apakah pilihannya adalah sekolah tradisional atau kelas yang diawasi secara khusus, itu tidak hanya mungkin, tetapi juga penting. Dengan bahan yang tepat dan dukungan orang tua, guru, dan seluruh komunitas pendidikan, pembelajaran membaca dan menulis juga dapat mereka akses, tergantung pada potensi pribadi mereka.
Peningkatan risiko penyakit tertentu
Orang dengan sindrom Down lebih rentan terhadap infeksi. Mereka lebih sering menderita kelainan jantung, organ, pencernaan atau perut. Mereka juga lebih rentan terhadap masalah THT, epilepsi atau penyakit autoimun tertentu.
Mereka mungkin juga lebih terpapar patologi seperti diabetes, hiper atau hipotiroidisme.
Salah satu elemen kunci dalam memantau orang dengan sindrom Down adalah gangguan penglihatan. Dengan demikian, pada populasi umum kasus katarak terjadi pada usia sekitar 70 tahun, sedangkan pada penderita Down Syndrome dapat terjadi pada usia 40 tahun.
Selain perawatan medis yang ketat, dukungan psikologis mungkin direkomendasikan. Penting, untuk kesejahteraan orang dengan sindrom Down dan orang-orang di sekitarnya, tidak diisolasi.
Trisomi autosomal
Down Syndrome dapat memengaruhi kromosom mana pun; tetapi ukuran kromosom ekstra menjadi faktor risiko keguguran, oleh karena itu kami terutama menjumpai trisomi 21 yang merupakan kromosom manusia terkecil. Trisomi 13 dan 18 juga dapat menyebabkan bayi lahir cukup bulan, tetapi anak tersebut paling sering meninggal dalam beberapa minggu2. Kasus lainnya hanya dapat bertahan dengan trisomi mosaik.
kromosom 1: trisomi mozaik 1 adalah bentuk yang sangat langka3
kromosom 2: trisomi mosaik 2 adalah bentuk langka dengan fenotipe4 yang sangat bervariasi
kromosom 3: mosaik trisomi 3 adalah bentuk yang langka5
kromosom 4: mosaik trisomi 4 menyebabkan retardasi pertumbuhan, defisit intelektual ringan, kelainan jantung, dysmorphia wajah, malformasi ibu jari dan kelainan kulit6
kromosom 5: trisomi mosaik 5 dapat asimtomatik atau menyebabkan berbagai cacat lahir7
kromosom 6: mosaik trisomi 6 adalah salah satu bentuk yang paling langka (7 kasus yang diketahui) 8
kromosom 7: mosaik trisomi 7 adalah bentuk langka dengan fenotipe yang sangat bervariasi9
kromosom 8: trisomi 8 menyebabkan sindrom Warkany
kromosom 9: trisomi 9 sangat jarang, beberapa kasus trisomi lengkap telah dijelaskan10
kromosom 10: Trisomi 10 mosaik biasanya berakibat fatal pada masa bayi11
kromosom 11: trisomi mosaik 11 hanya didokumentasikan dalam 3 kasus12
kromosom 12: Trisomi mosaik 12 merupakan bentuk langka yang dapat menyebabkan berbagai kelainan13
kromosom 13: trisomi 13 menyebabkan sindrom Patau
kromosom 14: trisomi mosaik 14 adalah bentuk langka dengan fenotipe yang sangat bervariasi14
kromosom 15: trisomi mosaik 15 menyebabkan retardasi pertumbuhan intrauterin dan kelainan jantung dan kraniofasial15
kromosom 16: trisomi mosaik 16 adalah bentuk langka dengan fenotipe variabel16
kromosom 17: trisomi mosaik 17 adalah bentuk yang langka, presentasi klinisnya sangat bervariasi17
kromosom 18: trisomi 18 menyebabkan sindrom Edwards
kromosom 19: trisomi mozaik 19 hanya diketahui pada 4 kasus18
kromosom 20: trisomi mosaik 20 adalah bentuk langka yang biasanya asimtomatik19
kromosom 21: trisomi 21 menyebabkan sindrom Down
kromosom 22: trisomi mosaik 22 adalah bentuk langka yang dapat menyebabkan berbagai manifestasi20
Rumus kromosom dari trisomi homogen autosom berbentuk 47, Xa, + b dengan:
a: X untuk anak perempuan dan Y untuk anak laki-laki
b: jumlah kelebihan kromosom
Misalnya 47, XY, + 21 untuk trisomi 21 pada anak laki-laki21. Untuk trisomi mozaik, garis miring ditambahkan diikuti dengan rumus kromosom normal, trisomi mozaik 10 akan ditandai dengan 47, XX, + 10/46, XX.
Prinsip
Trisomi biasanya hasil dari nondisjunction. Non-disjungsi dapat terjadi selama gametogenesis ibu atau ayah (pada manusia, sebagian besar berasal dari ibu). Ini adalah kesalahan pembelahan sel dimana terjadi kegagalan pasangan kromosom untuk berpisah. Ini menghasilkan distribusi yang tidak merata dari sepasang kromosom yang homolog ke sel anak: seluruh pasangan kromosom berpindah ke salah satu sel anak dan sel anak lainnya tidak menerima apa-apa.
Setelah pembuahan, sel dengan satu pasangan kromosom menerima kromosom ketiga dari gamet lainnya, sehingga menjadi zigot dengan sindrom Down (ketika sel anak lainnya dibuahi, ini menyebabkan monosomi).
Jika nondisjungsi terjadi setelah pembuahan, diperoleh trisomi mozaik.
Terkadang zigot sindrom Down sembuh sendiri dengan kehilangan salah satu dari tiga kromosom ekstra (reduksi trisomi), yang dapat menyebabkan disomi uniparental ketika dua kromosom yang dipertahankan berasal dari induk yang sama.
Epidemiologi
Sindrom Down mempengaruhi sejumlah besar kehamilan, risikonya meningkat seiring bertambahnya usia ibu: dari sekitar 2% kehamilan pada usia 25 tahun, hingga 35% pada 40 tahun24; sebagian besar kehamilan ini akan berakhir dengan keguguran. Trisomi 16 adalah yang paling sering ditemui, diperkirakan terjadi pada 1 sampai 1,5% kehamilan. Trisomi 15 dan 22 adalah dua bentuk umum lainnya, mereka juga menyebabkan kematian dalam rahim janin yang terkena. Ada kemungkinan bahwa hasil ini bias karena kegagalan mendeteksi kegagalan kehamilan yang sangat dini untuk trisomi tertentu; Faktanya, frekuensi relatif dari trisomi yang terdeteksi oleh diagnosis praimplantasi lebih homogen.
Di antara kehamilan yang layak; trisomi yang paling sering adalah trisomi 21 atau sindrom Down, diperkirakan 1/800 kelahiran tanpa diagnosis prenatal28. Trisomi lain dari autosom lebih jarang: 1/8000 kelahiran untuk trisomi 18; 1 / 100.000 untuk trisomi 13. Namun, sebagian besar kehamilan gagal secara spontan: pada 75% kasus untuk trisomi 21 30 dan lebih dari 95% untuk trisomi 18 dan 1331.
Duplikasi kromosom seks cukup umum. Trisomi X mempengaruhi 1/1000 kelahiran pada anak perempuan; Sindrom XXY 1/500 kelahiran anak laki-laki33 dan 1/1000 untuk bentuk XYY34.
Evolusi
Anak-anak dengan sindrom Down 13 dan 18 umumnya hanya hidup dari beberapa hari hingga beberapa minggu karena pentingnya kelainan yang terkait.
Sindrom Down mengganggu perkembangan otak, menyebabkan defisit kognitif dan gangguan perilaku. Namun, dengan stimulasi, orang dengan sindrom Down dapat berkembang secara intelektual dan emosional serta profesional, bahkan jika kecacatan mereka tidak akan pernah memungkinkan mereka mencapai usia mental dewasa.
Sindrom yang disebabkan oleh trisomi kromosom seks tidak terlalu parah dan bahkan seringkali asimtomatik. Mereka terutama dimanifestasikan oleh masalah dengan perkembangan seksual atau kesuburan. Mekanisme perlindungan mungkin terkait dengan inaktivasi kromosom X dan rendahnya jumlah gen yang dibawa oleh kromosom Y2.
Sindrom Edwards, juga disebut trisomi 18
Sindrom Edwards, juga disebut trisomi 18, adalah penyakit kromosom bawaan yang disebabkan oleh adanya kromosom ekstra untuk pasangan ke-18. Sindrom malformasi ini biasanya berujung pada kematian dini. Penyakit ini dijelaskan oleh ahli genetika Inggris John H. Edwards dalam artikel tahun 1960.
Penyebab
Sebagian besar kasus trisomi 18 terjadi karena adanya tiga salinan kromosom 18 di setiap sel tubuh, bukan dua salinan biasa. Materi genetik tambahan mengganggu jalannya perkembangan normal, menyebabkan ciri-ciri khas sindrom Down.
Sekitar 5% orang dengan Down Syndrome memiliki salinan ekstra kromosom 18 hanya di sel-sel tertentu di tubuh. Pada orang-orang ini, kondisi tersebut disebut trisomi mosaik 18. Tingkat keparahan mozaik trisomi 18 tergantung pada jenis dan jumlah sel yang memiliki kromosom ekstra. Perkembangan individu dengan bentuk trisomi 18 ini dapat berkisar dari normal hingga sangat terpengaruh.
Sangat jarang, bagian lengan panjang (q) kromosom 18 menempel pada kromosom lain selama pembentukan sel reproduksi (telur dan sperma) atau sangat awal dalam perkembangan embrio. Individu yang terkena memiliki dua salinan kromosom 18, ditambah bahan ekstra dari kromosom 18 yang menempel pada kromosom lain. Orang dengan perubahan genetik ini akan mengalami trisomi parsial 18. Jika hanya sebagian dari lengan q hadir dalam rangkap tiga, tanda fisik dari trisomi parsial 18 mungkin kurang parah daripada yang biasanya terlihat pada trisomi 18. Jika semua lengan q adalah hadir dalam rangkap tiga, individu dapat terkena dampak parah seolah-olah mereka memiliki tiga salinan lengkap kromosom 18.
Mode transmisi
Kebanyakan kasus trisomi 18 tidak turun-temurun, tetapi terjadi sebagai kejadian acak selama pembentukan sel telur dan sperma. Kesalahan dalam pembelahan sel yang disebut nondisjunction menghasilkan sel reproduksi dengan jumlah kromosom yang tidak normal. Misalnya, sel telur atau sel sperma dapat memperoleh salinan ekstra kromosom 18. Jika salah satu dari sel reproduksi atipikal ini berkontribusi pada susunan genetik anak, anak tersebut akan memiliki kromosom ekstra 18 di setiap sel dalam tubuh.
Trisomi 18 mosaik juga tidak diwariskan. Itu terjadi sebagai peristiwa acak selama pembelahan sel pada awal perkembangan embrio. Akibatnya, beberapa sel dalam tubuh biasanya memiliki dua salinan kromosom 18, dan sel lain memiliki tiga salinan kromosom ini.
Trisomi parsial 18 bisa diturunkan. Orang yang tidak terpengaruh dapat mengatur ulang materi genetik antara kromosom 18 dan kromosom lain. Penataan ulang ini disebut translokasi seimbang karena tidak ada bahan tambahan dari kromosom 18. Meskipun tidak menunjukkan tanda-tanda trisomi 18, orang yang membawa jenis translokasi seimbang ini berisiko lebih tinggi. Memiliki anak dengan penyakit tersebut.
Anomali dicatat
Orang dengan trisomi 18 sering mengalami pertumbuhan lambat sebelum lahir (retardasi pertumbuhan intrauterin) dan berat badan lahir rendah. Orang yang terkena mungkin memiliki kelainan jantung dan kelainan organ lain yang berkembang sebelum lahir. Ciri-ciri lain sindrom Down termasuk kepala kecil berbentuk tidak normal; rahang kecil dan mulut; dan kepalan tangan terkepal dengan jari yang tumpang tindih. Karena adanya beberapa kondisi medis yang mengancam jiwa, banyak orang dengan sindrom Down meninggal sebelum lahir atau selama bulan pertama mereka. Lima sampai 10 persen anak-anak dengan penyakit ini hidup melewati tahun pertama mereka, dan anak-anak ini seringkali menderita cacat intelektual yang parah.
Anak-anak dengan kondisi tersebut biasanya hanya bertahan beberapa minggu dan tidak lebih dari setahun. Ada beberapa kasus pasien yang selamat setidaknya sampai usia 19 tahun. Terlepas dari semuanya, ini lebih jarang daripada trisomi 21, yang merupakan trisomi yang paling umum dan paling layak. Namun, seperti trisomi 13 jauh lebih serius daripada trisomi 21 karena sebagian besar kasus meninggal dalam rahim sebelum 6 bulan, karena trisomi 18 ini mencegah perkembangan bayi baru lahir.
Dampak
Ini adalah kelainan kromosom langka yang mempengaruhi sekitar satu dari 6.000 orang.
Tanda klinis
Dysmorphia kraniofasial
Dolichocephaly (oksiput menonjol dan TID pendek)
Mulut kecil, mikrognatia
Telinga fauna: paviliun tidak terlalu sempit, datar, runcing di bagian atasnya.
Leher-Dada-Perut
Leher pendek
Panggul yang kencang
Anggota
Ciri khas tangan: kepalan tangan tertutup, jari telunjuk menutupi jari tengah, jari kelingking menutupi jari manis.
Sikap pemohon – kapak es.
Malformasi
Jantung: CIV (komunikasi interventrikular), CA, CIA …
Paru, gastrointestinal, ginjal…
Trisomi 13, atau sindrom Patau
Trisomi 13, atau sindrom Patau, adalah kondisi yang disebabkan oleh adanya tambahan kromosom 13. Oleh karena itu, rumus kromosom pasien adalah 47 kromosom, bukan 46 kromosom pada manusia. Klaus Patau adalah orang pertama yang mendeskripsikan trisomi 132 pada tahun 1960.
Meskipun trisomi 13 adalah yang paling langka dari trisomi yang dapat menyebabkan bayi lahir cukup bulan, 3 ini juga merupakan kelainan kromosom yang paling sering yang ditandai dengan beberapa malformasi dan hanya menyisakan sedikit harapan untuk bertahan hidup. Setelah diagnosis 1. Patologi ini memengaruhi sejumlah besar organ. Pada tahun 1960 Patau menemukan kromosom ekstra. Kelangsungan hidup sampai dewasa dimungkinkan, terutama dalam kasus mozaik.
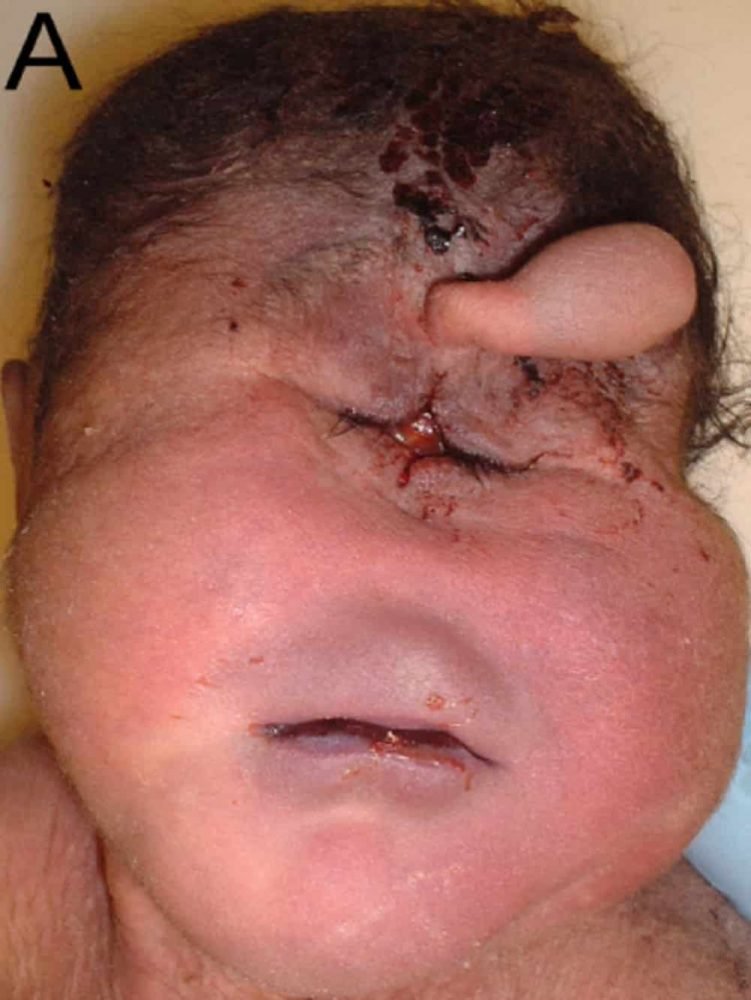
Deskripsi
Janin mengalami retardasi pertumbuhan intrauterin dengan setidaknya beberapa gejala berikut:
Kelainan sistem saraf
Holoproencephaly adalah malformasi yang paling umum (50%)
Dilatasi sambungan ventrikel
Pembesaran fossa posterior
Kelainan wajah
Penurunan jarak antar orbital (hipotelorisme) hingga hanya ada satu mata yang mencapai aspek cyclops
Divisi labio-palatal
Kelainan ginjal
Hidronefrosis
Peningkatan ukuran ginjal
Kelainan jantung
Komunikasi interventrikuler
Displasia katup
Tetralogi Fallot
Kelainan tungkai
Polydactyly
Kaki pengkor
Kelainan perut
Omphalocele
Ekstrofi kandung kemih
Kariotipe dengan amniosentesis, sampel darah janin, atau biopsi trofoblas membuat diagnosis.
Perbedaan diagnosa
Penting untuk membuat diagnosis yang tepat untuk konseling genetik yang paling tepat pada orang tua.
Penyakit kromosom ini harus dibedakan dari kelainan genetik berikut:
- Sindrom Cohen-Gorlin (Kelainan langka perkembangan embrio yang ditandai dengan mikrosefali, dysmorphia wajah, hipotonia, disabilitas intelektual non-progresif, miopia, distrofi retinal, neutropenia, dan obesitas tubuh).
- Sindrom Meckel-Gruber (sindrom polimalformasi yang jarang terjadi, transmisi resesif autosomal, yang didefinisikan oleh ensefalokel oksipital, polidaktili, dan displasia kistik ginjal. Ultrasonografi saat ini merupakan cara terbaik untuk skrining antenatal untuk polimalformasi yang mematikan ini dan konfirmasinya dilakukan dengan mempelajari kariotipe. Kami melaporkan kasus sindrom Meckel yang ditemukan dengan USG. Kehamilan diakhiri pada usia kehamilan 25 minggu).
- Sindrom Smith-Lemli-Opitz (ditandai dengan banyak cacat lahir, cacat intelektual dan gangguan perilaku).
Kematian
80 sampai 90% janin dengan trisomi 13 meninggal dalam rahim. Separuh dari anak-anak yang mencapai cukup bulan meninggal dalam waktu tiga bulan setelah kelahiran mereka.
Konseling genetik
Karena risiko kekambuhan sangat tinggi, disarankan untuk melakukan kariotipe dengan sampel trofoblas pada kehamilan berikutnya dari 12 minggu kehamilan, sampel ini adalah BVC (biopsi vilus korionik). Hanya risiko trisomi 13 yang meningkat, risiko kelainan kromosom lainnya tetap tidak berubah.
Trisomi 9
Trisomi 9 adalah penyakit kromosom kongenital yang disebabkan adanya ekstra kromosom untuk pasangan ke-9.
Bentuk lengkap dan homogen sangat jarang, menyebabkan aborsi spontan pada 80% kasus, dan sebaliknya menyebabkan kematian dalam beberapa jam setelah lahir1.
Bentuk mosaik relatif lebih jarang (200 kasus dijelaskan dalam literatur) dan dapat bertahan, konsekuensinya tergantung pada proporsi sel yang terpengaruh. Sindrom ini pertama kali dijelaskan pada tahun 1970 oleh ahli genetika Prancis Marie-Odile Rethoré.
Gejala
Adanya materi genetik tambahan akibat trisomi parsial 9 p menyebabkan berbagai gejala pada orang dengan sindrom Rethoré, meskipun tidak semua fitur atau tidak semua fitur hadir pada seseorang pada tingkat yang sama. Beberapa ciri fisik sudah dapat dikenali selama pemeriksaan USG sebagai bagian dari diagnosa prenatal. Fitur yang paling umum meliputi:
Cacat jantung
Bintik putih (fenomena bola golf di hati)
Retardasi perkembangan motorik
Keterlambatan pematangan tulang
Perawakan pendek (pertumbuhan panjang di bawah rata-rata)
Microcephaly (kepala yang relatif kecil) dengan pertumbuhan berlebih vertikal (skaphocephalus / tengkorak panjang), tetapi masih kepala yang cukup lebar
Malformasi serebelar
Pembesaran ventrikel otak
Malformasi Dandy Walker
jarak interpupillary yang relatif kecil (hipotelorisme) dan mata yang relatif kecil (microphthalmia)
tinggi, dahi melengkung
hidung pendek dan lebar
telinga dalam, kadang-kadang berubah bentuk (displastik)
lipatan kecil berbentuk bulan sabit di sudut dalam mata (epicanthus medialis)
Invaginasi tepi kelopak mata
sumbu kelopak mata miring ke luar (sudut luar kelopak mata lebih rendah dari sudut dalam kelopak mata)
Bola mata tenggelam ke dalam rongga mata dengan penglihatan yang berkurang (enophthalemia / nanophthalmos)
retrognathia mandibula = perpindahan rahang bawah ke belakang
Bibir dan langit-langit sumbing
Malformasi tabung saraf
Ciri-ciri khusus kuku (misalnya pemutihan total pada kuku akibat retensi udara, juga dapat muncul sebagai bintik seukuran kepala jarum, garis horizontal berbentuk bulan sabit, atau garis horizontal lebar bergantian)
posisi khusus jari dan / atau kaki (membungkuk / dalam posisi fleksi)
Malformasi di area saluran urogenital (termasuk semua organ yang digunakan untuk pembentukan dan ekskresi urin atau reproduksi seksual, misalnya organ genital internal dan eksternal, ginjal, dll. / Pada anak laki-laki dengan trisomi 9, untuk Misalnya sering tertinggal salah satu atau kedua testis di rongga perut atau di kanalis inguinalis / kriptorkismus)
Hernia diafragma (perforasi diafragma)
Penumpukan kalsium di hati
arteri umbilikalis tunggal (arteri umbilikalis tunggal)
sebagian besar cacat kognitif yang parah
Namun, tidak satu pun dari gejala ini yang cukup meyakinkan untuk diagnosis yang jelas, bahkan jika beberapa fitur khusus muncul bersamaan satu sama lain.
Diagnosa
Diagnosis masih hanya mungkin dilakukan dengan memeriksa kromosom itu sendiri. Metode prenatal khususnya amniosentesis atau pengambilan sampel vilus korionik atau analisis kromosom yang mengikuti prosedur ini.
Sindrom Pätau (trisomi 13), sindrom Edwards (trisomi 18), sindrom Wolf-Hirschhorn dan triploidi adalah diagnosis banding yang mungkin.
Prognosis dan terapi
Tidak ada bentuk trisomi 9 yang dapat disembuhkan secara kausal. Oleh karena itu, terapi hanya mungkin selama gejala tertentu oleh medis – terapi dapat mengurangi atau mengimbangi metode. Sejauh ini, banyak anak dengan trisomi 9 meninggal selama kehamilan atau dalam waktu yang relatif singkat setelah lahir. Pada anak-anak yang hidup lebih lama, mosaik trisomi 9 biasanya dapat ditemukan dan, tergantung pada proporsi sel-sel disomer, prognosisnya seringkali lebih baik, di mana itu juga tergantung pada karakteristik fisik mana yang ada di mana manifestasi dan bagaimana mereka dapat dirawat. atau bagaimana mereka diperlakukan. Gangguan kognitif umumnya diklasifikasikan sebagai parah.
Sindrom Warkany, juga disebut trisomi 8
Sindrom Warkany, juga disebut trisomi 8, adalah penyakit kromosom bawaan yang disebabkan oleh adanya kromosom ekstra untuk pasangan ke-8.
Penderita penyakit trisomi 8 mozaik dapat hidup seperti penderita trisomi 21, dengan gejala dan kelainan fisik yang kurang lebih sama1,2, sedangkan janin dengan trisomi 8 komplit sangat jarang lahir dan meninggal dalam kandungan.
Seperti trisomi 21 dan trisomi 13, risikonya meningkat seiring dengan usia ibu.
Frekuensi terjadinya
Trisomi 8 adalah salah satu keanehan kromosom yang langka. Terjadi secara sporadis (sporadis, random), sekitar 120 kasus telah terdokumentasi. Trisomi 8 didominasi oleh mozaik (mozaik trisomi 8 / trisomi 8 sindrom mozaik). Baik anak laki-laki maupun perempuan bisa lahir dengan trisomi 8.
Tanda-tanda umum sebelum lahir (sebelum lahir)
Dalam perjalanan kemungkinan pemeriksaan prenatal yang terus berkembang (diagnosis prenatal), beberapa keanehan telah didokumentasikan yang dapat ditemukan relatif sering pada bayi yang belum lahir dengan trisomi 8.
Tanda-tanda yang dapat menunjukkan adanya trisomi 8 pada janin, terutama kombinasi satu sama lain, dan kadang-kadang dapat dikenali melalui pemeriksaan USG, antara lain:
Cacat jantung
Malformasi ginjal, pembesaran panggul ginjal
Malformasi kerangka (tetramellia)
Membengkokkan tulang belakang dengan lengkungan ke luar ke depan (skoliosis, lordosis, kyphosis)
dua atau lebih vertebra yang berurutan seluruhnya atau sebagian bergabung bersama (blok vertebra)
sedikit menekuk jari kelingking ke arah jari manis (secara klinis) dengan pemendekan tendon dan selubung tendon secara bersamaan, yang membuat ekstensi lengkap dari masing-masing jari tidak mungkin (camptodactyly)
daerah mulut-dagu yang relatif kecil (retrognati mandibula) dengan pemendekan rahang bawah
Jari pendek (brachydactyly)
sendok iliaka sempit
Hilang atau malformasi tempurung lutut (aplasia atau hipoplasia patela)
Penumpukan cairan di area leher (transparansi leher tinggi)
Namun, tidak satu pun dari gejala ini yang cukup meyakinkan untuk diagnosis yang jelas, bahkan jika beberapa fitur khusus muncul bersamaan satu sama lain.
Sampai saat ini, diagnosis hanya dapat dilakukan dengan memeriksa kromosomnya sendiri. Metode prenatal khususnya amniosentesis atau pengambilan sampel vilus korionik atau analisis kromosom yang mengikuti prosedur ini. Khususnya dengan pengambilan sampel vilus korionik, harus diingat bahwa trisomi mosaik 8 juga dapat terjadi di plasenta, dan bayi tidak memiliki mosaik janin dan mosaik tidak selalu dikenali selama pemeriksaan kromosom lainnya.
Tanda umum setelah lahir (postnatal)
Setelah lahir, sebagian besar bayi dengan trisomi 8 memiliki karakteristik fisik berbeda yang memungkinkan diagnosis yang dicurigai. Ini termasuk:
kekakuan sendi bawaan (arthrogryposis)
sering spina bifida occulta
Ciri-ciri khusus telapak kaki (terutama lekukan yang terlihat pada telapak kaki / alur plantar)
jarak yang relatif jauh antara jari kaki pertama dan kedua (celah sandal / alur sandal)
banyak dan dalam alur di telapak tangan (alur telapak tangan)
Alur empat jari (sekitar 75 dari 100 anak)
telinganya relatif dalam, besar dan panjang
lipatan terbelakang (hipoplastik) yang terletak di seberang tepi daun telinga (antheliks)
dahi tinggi
leher pendek
bahu sempit dan miring
dada panjang
Ciri khusus dari jumlah dan lebar tulang rusuk
kelebihan (aksesori) puting (puting)
tinggi, langit-langit sempit
Langit-langit sumbing
bibir penuh, montok, bibir bawah terbuka
bantuan mata yang relatif kecil (hipotelorisme)
Satu atau kedua kelopak mata terkulai, dengan kelopak mata bagian atas lebih mungkin terpengaruh (ptosis)
Juling (strabismus)
hidung lebar, sering lubang hidung ke atas (nares)
seringkali di atas berat rata-rata
peningkatan risiko tumor
cacat kognitif ringan sampai sedang
Sindrom lain dengan arthrogryposis dapat digunakan sebagai diagnosis banding.
Sejarah
Trisomi 8 tidak bisa disembuhkan secara kausal, hanya gejalanya yang bisa diobati. Gambaran klinisnya cukup bervariasi, terutama pada trisomi mosaik 8, dan orang-orang juga diketahui hampir tidak memiliki kelainan apa pun.
Prognosis untuk perkembangan fisik bergantung pada karakteristik fisik mana yang muncul dalam bentuk apa dan bagaimana mereka dapat dirawat atau bagaimana mereka dirawat.
Gangguan kognitif umumnya diklasifikasikan sebagai ringan sampai sedang, pada orang dengan trisomi mosaik 8 seringkali lebih ringan dan sangat bervariasi dalam tingkat keparahan. Dengan proporsi sel trisomerik yang jauh lebih rendah, dimungkinkan juga untuk mencapai kecerdasan biasa.
Down Syndrome mempengaruhi kromosom seks
Karena trisomi ditentukan oleh adanya kromosom ekstra dalam kariotipe, semua kromosom dapat terlibat, termasuk kromosom seks. Juga ada trisomi yang mempengaruhi pasangan kromosom X atau XY. Konsekuensi utama dari trisomi ini adalah mempengaruhi fungsi kromosom seks, termasuk kadar hormon seks dan organ reproduksi.
Ada tiga jenis trisomi kromosom seks:
- XXX (triple X)
- XXY (Klinefelter syndrome or 47)
- XYY (“double Y” or Syndrome 47)
(A) Sindrom Triple X
Sindrom Triple X adalah aneuploidi kromosom seks pada wanita. Ini ditandai dengan adanya kromosom X tambahan di setiap sel wanita (karena itu homogen). Sindrom ini juga disebut sindrom XXX, trisomi X, triplo-X, 47xxx atau aneuploidy 47, XxxxX. Varian kromosom ini dihasilkan dari produksi gamet diploid selama meiosis.
Trisomy-X muncul selama pembagian gamet orang tua. Sekitar 1 dari 1.000 wanita terpengaruh. 10% wanita yang didiagnosis (di antara mereka dengan trisomi-X) dan yang menunjukkan gejala biasanya lebih besar dari rata-rata.
Gejala
Seringkali, trisomi X tidak menyebabkan masalah medis atau ciri-ciri tertentu, dan tidak terdeteksi. Wanita dengan sindrom ini umumnya lebih tinggi dari rata-rata, mungkin memiliki siklus menstruasi yang tidak teratur, dan berisiko lebih besar mengalami kesulitan belajar, terutama kesulitan bahasa, jarang termasuk keterbelakangan mental. Mereka dapat menunjukkan ketidakstabilan emosional.
Para peneliti belum sepenuhnya memahami hubungan yang jelas antara salinan ekstra kromosom X dan kesulitan belajar pada beberapa anak perempuan dan perempuan, tetapi ditetapkan bahwa ukurannya yang besar disebabkan oleh keberadaan gen SHOX (gen yang bertanggung jawab); hadir di lengan pendek X, dan oleh karena itu di sini dalam 3 salinan) yang kemudian diekspresikan secara berlebihan. Sekitar 10% dari wanita yang terkena mengalami masalah ginjal. Namun, karakteristik ini sangat bervariasi antara individu yang terkena. Di sisi lain, kebanyakan wanita dengan sindrom triple X memiliki perkembangan seksual yang normal dan mampu mengandung anak yang sehat.
Penyebab
Disebabkan seperti sebagian besar trisomi sebagai akibat dari distribusi kromosom yang buruk selama gametogenesis, distribusi yang buruk ini dapat terjadi baik selama oogenesis (anafase I atau anafase II) dan selama spermatogenesis (hanya anafase II, ketika kromosom X ganda ayah terpecah menjadi dua kromosom X tunggal dan kedua salinannya dimulai dari sisi sel yang sama). Seperti trisomi lainnya atau semua aneuploidi, kejadiannya meningkat seiring dengan usia ibu.
(B) Sindrom Klinefelter atau 47, XXY
Sindrom Klinefelter atau 47, XXY adalah aneuploidi yang ditandai pada manusia dengan tambahan kromosom seks X. Individu kemudian menampilkan dua kromosom X dan satu kromosom Y, yaitu 47 kromosom, bukan 46. Individu tersebut kemudian bersifat laki-laki, tetapi tidak subur. Rumus kromosomnya tertulis “47, XXY”.
Sindrom ini dijelaskan pada tahun 1942 tetapi etiologinya tidak diketahui. Pada tahun 1959, asal kromosom dari sindrom ini ditemukan.
Penyebab
Asal-usul keanehan ini ditemukan selama meiosis gamet orang tua subjek, ketika kromosom seks tidak terdistribusi secara normal. Kelompok lain yang disebut “mosaik” menemukan asalnya selama mitosis. Kelompok ini menyumbang sekitar 10% dari kasus sindrom Klinefelter. Individu yang disebut “mosaik” tidak memiliki semua sel yang dipengaruhi oleh karakteristik kromosom dan oleh karena itu untuk beberapa dapat memiliki anak.
Frekuensi
Banyak orang membawa kariotipe ini tanpa benar-benar terpengaruh (terlepas dari ketidaksuburannya). Sekitar satu dari setiap 500 hingga 1.000 kelahiran laki-laki adalah pembawa sindrom ini. Kariotipe 47, XXY (80% kasus Klinefelter) harus dibedakan dari kariotipe 48, XXXY, 48, XXYY dan 49, XXXXY dan mosaik lain yang kemudian menghadirkan konsekuensi selain yang disebutkan di atas dan yang merupakan 20% kasus.
Kurang dari 10% dari sindrom ini didiagnosis sebelum dewasa dan kemungkinan hanya seperempat kasus yang benar-benar terdeteksi.
Gejala
Di bawah nama sindrom Klinefelter kami mengelompokkan semua atau sebagian dari semua gejala berikut, variabilitas ekspresi yang sering diamati dan semua masalah hidup yang tidak dapat dikaitkan dengan sindrom ini: ukuran rata-rata lebih besar dari saudara kandung, kemungkinan penundaan pada masa pubertas, kemungkinan selama masa kanak-kanak mengalami ketidakmampuan belajar bahasa atau membaca, ukuran testis lebih kecil dari masa pubertas, kemungkinan pada masa remaja jika ada kekurangan testosteron dari rambut yang rendah, kekurang tonjolan otot, perkembangan kelenjar susu atau ginekomastia, rapuh email gigi dan osteoporosis di usia dewasa.
Oleh karena itu, ekspresi atipikal dari sindrom ini menjelaskan seringnya keterlambatan dalam diagnosisnya, yang seringkali dilakukan hanya sebagai bagian dari pencarian kemandulan.
Diagnostik
Biasanya terdapat gambaran biologis hipogonadisme hipergonadotropik, dengan, pada orang dewasa, konsentrasi testosteron normal atau rendah dan peningkatan kadar LH dan FSH5.
Diagnosis dibuat dengan menggunakan kariotipe tetapi jauh lebih sulit jika terjadi mosaik.
Komplikasi
Kematian secara keseluruhan meningkat, terutama dari penyebab kardiovaskular, neurologis, atau paru.
Risiko kanker payudara meningkat serta risiko penyakit tromboemboli, diabetes atau osteoporosis.
Perawatan
Untuk anak-anak dengan kesulitan belajar, pembentukan perawatan awal dan tradisional dalam terapi wicara dan terapi okupasi dapat membantu.
Perawatan hormonal dengan testosteron sejak pubertas mungkin ditawarkan. Ini memperbaiki gejala (tanpa memecahkan masalah ketidaksuburan) dan dapat mencegah beberapa komplikasi lanjut seperti osteoporosis.
Jika terjadi ginekomastia yang signifikan dan melumpuhkan, operasi plastik mungkin ditawarkan. Demikian juga, seseorang dapat melakukan eksisi pada testis kecil pada mereka yang mengalami kesulitan menerima kondisi ini, dengan pemasangan prostesis testis.
Penatalaksanaan infertilitas dilakukan oleh tim Medical Assisted Prokreasi (AMP), yang akan menentukan derajat infertilitas dan berbagai kemungkinan yang ditimbulkannya (fertilisasi in vitro dan injeksi sperma intrasitoplasma; inseminasi buatan dengan donor; adopsi). Azospermia (tidak adanya spermatozoa saat ejakulasi) terkadang bukan merupakan saksi dari tidak adanya produksi sperma dan sampel intratesticular telah diambil.
Rekomendasi
Akademi andrologi Eropa menerbitkan rekomendasi tentang sindrom Klinefelter pada tahun 2020.
(C) Sindrom 47, XYY (“Y ganda”)
Sindrom 47, XYY (“Y ganda”) adalah aneuploidi manusia, lebih khusus lagi trisomi yang dijelaskan oleh adanya abnormalitas kromosom Y kedua. Biasanya kariotipe manusia terdiri dari 46 kromosom atau 23 pasang, pada kasus sindrom ini terdapat kromosom 2×23 + 1 (2n + 1), sesuai dengan trisomi yang disebut disomi Y (adanya 2 kromosom Y).
Penggunaan istilah “sindrom” dipertanyakan oleh beberapa ahli genetika. Mereka yang terkena memang memiliki fenotipe laki-laki normal dan banyak dari mereka bahkan tidak mengetahui kariotipe mereka.
Sindrom ini juga disebut sindrom Yakub atau sindrom pembunuh.
Ciri-ciri fisik
Sebagian besar waktu, kehadiran kromosom Y ekstra tidak disertai dengan ciri fisik atau masalah medis yang tidak biasa. Di masa kanak-kanak, anak-anak mengalami percepatan pertumbuhan dengan, rata-rata, tinggi akhir yang akan menjadi 7 cm lebih besar dari yang diharapkan. Beberapa kasus jerawat parah telah terdeteksi tetapi ahli kulit yang mengkhususkan diri pada masalah jerawat sekarang mempertanyakan keberadaan hubungan dengan 47-XYY.
Kadar testosteron prenatal dan postnatal normal. Banyak orang dengan sindrom ini memiliki perkembangan seksual dan kesuburan pria yang normal. Karena sindrom XYY tidak dicirikan oleh ciri-ciri fisik yang khas, kelainan tersebut seringkali hanya dicatat selama analisis genetik dilakukan untuk tujuan lain.
Karakteristik perilaku
Anak-anak dengan 47, sindrom XYY memiliki risiko lebih tinggi mengalami masalah belajar (50% lebih tinggi) dan perkembangan bahasa yang tertunda. Sebaliknya, survei nasional yang dilakukan pada tahun 2004 di Amerika Serikat oleh “Center for Disease Control and Prevention” menunjukkan bahwa 10% anak yang terkena cacat genetik ini mengalami masalah belajar.
Untuk anak-anak dengan kelainan 47, XXY (laki-laki) dan 47, XXX (perempuan), nilai tes IQ anak-anak dengan sindrom 47, XYY rata-rata antara 10 dan 15 poin di bawah hasil saudara dan saudara perempuan mereka. Namun, perbedaan ini (rata-rata 12 poin) adalah yang biasanya diamati antara anak-anak dari keluarga yang sama. Dari 14 kasus anak dari keluarga dengan latar belakang sosial ekonomi tinggi, IQ 6 di antaranya antara 100 dan 147 dengan rata-rata 120. Di antara 11 anak yang memiliki saudara kandung, 9 memiliki saudara kandung yang berprestasi lebih baik di sekolah, tetapi dalam satu dalam kasus hasil mereka sama dan dalam kasus terakhir mereka bahkan lebih rendah daripada anak.
Keterlambatan perkembangan atau masalah perilaku juga dapat diamati dan orang-orang ini cenderung lebih agresif. Tetapi karakteristik ini memiliki variabilitas yang besar pada populasi yang terkena, tidak spesifik untuk orang dengan 47, sindrom XYY, dan tidak diekspresikan secara berbeda dari orang dengan kariotipe laki-laki 46, XY yang khas.
Penyebab
D47, XYY bukan merupakan penyakit turunan, anomali muncul secara acak selama pembentukan sel germinal. Disjungsi gonosom yang tidak tepat selama anafase II meiosis dapat menyebabkan sel germinal muncul dengan salinan ekstra kromosom Y. Jika salah satu sperma atipikal ini terlibat dalam pembuahan, anak akan memiliki kromosom Y ekstra di semua selnya.
Dalam beberapa kasus, kesalahan terjadi pada tingkat pembelahan sel mitosis postzygotic pada awal perkembangan embrio. Ini dapat menghasilkan mozaik antara 46, XY dan 47, XYY.
Frekuensi
Frekuensi observasi sindrom 47, XYY adalah 1 per 1000 kelahiran anak laki-laki1. Usia orang tua tidak berpengaruh terhadap frekuensi kemunculan.
Kasus pertama
Kasus terdokumentasi pertama seseorang dengan kariotipe 47, XYY dilaporkan oleh Avery A. Sandberg dan rekannya di Roswell Park Memorial Institute di Buffalo, New York, pada tahun 1961. Penemuan itu tidak disengaja dalam subjek berusia 44 tahun, 183 cm tinggi dan dengan kecerdasan normal, saat melakukan analisis kariotipe karena putrinya mengidap sindroma Down.
47, XYY adalah aneuploidi kromosom seks terakhir yang ditemukan (47, XXY dan 45, X ditemukan dua tahun sebelumnya dan 47, XXX pada tahun 1959). Bahkan kasus yang sangat jarang yaitu 48, XXYY telah terungkap setahun sebelumnya pada tahun 1960. Visualisasi aneuploidi yang mempengaruhi kromosom X dapat dicapai dengan tidak adanya atau adanya heterokromatin “betina” (sel Barr’s). Dalam inti sel interfase selama studi tentang apusan oral (teknik dikembangkan sepuluh tahun sebelum laporan aneuploidi seksual pertama).
Teknik analog untuk memvisualisasikan aneuploidi pada kromosom Y dengan kehadiran heterokromatin “jantan” yang berlebihan tidak dikembangkan sampai tahun 1970 (sepuluh tahun setelah aneuploidi pertama pada kromosom seks).
Sumber bacaan: Cleverly Smart, Health Line, CDC, Mayo Clinic
Sumber foto (utama): Uncomfortable Revolution
Pinter Pandai “Bersama-Sama Berbagi Ilmu”
Quiz | Matematika | IPA | Geografi & Sejarah | Info Unik | Lainnya | Business & Marketing